La Sindrome di Klinefelter: è importante una diagnosi precoce
Descritta nel 1942, la Sindrome di Klinefelter è una condizione genetica caratterizzata da testicoli piccoli, ginecomastia, ipogonadismo e azoospermia. La sua causa è stata identificata nel 1959 con la scoperta di un cromosoma X in più, classificandola tra le aneuploidie sessuali. Solo il 25% dei pazienti riceve una diagnosi dopo la nascita e meno del 10% prima della pubertà, un ritardo che limita le possibilità di intervento precoce, soprattutto per la spermatogenesi. Nuove evidenze scientifiche sottolineano l'importanza di una diagnosi tempestiva per migliorare i trattamenti disponibili.
Indice
Cos'è la Sindrome di Klinefelter?
La Sindrome di Klinefelter è stata descritta nel 1942 da Harry Klinefelter come un’entità clinica a sé stante, caratterizzata da ginecomastia, testicoli piccoli e duri, ipogonadismo, azoospermia ed aumentati livelli sierici di FSH.
La causa di questa patologia è rimasta sconosciuta sino al 1959, anno in cui fu dimostrata la presenza di un extracromosoma X nel cariotipo di un paziente con questa sindrome che venne così classificata per la prima volta tra le aneuploidie che interessano i cromosomi sessuali.
Epidemiologia
Da studi epidemiologici eseguiti in diversi paesi oggi è stata stimata una prevalenza pari a 152 casi per 100.000 maschi vivi o un caso su 660 con un’incidenza di 1-2 per 1000 neonati maschi vivi.
L’80% dei Klinefelter presenta come cariotipo 47,XXY ed il rimanente 20% presenta mosaicismi tipo 47,XXY/46,XY (cariotipi diversi in cellule diverse), X sovra numerari (48,XXY; 49,XXXXY), una o diverse Y in più (48,XXYY), o X sovra numerari e strutturalmente abnormi.
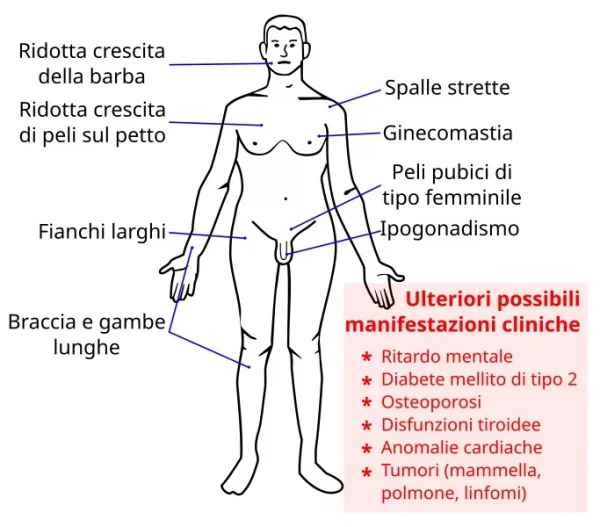
Le caratteristiche cliniche dei soggetti con Sindrome di Klinefelter differiscono a seconda dell’assetto del cariotipo e dell’età del paziente; oltre alle fisiologiche variazioni interindividuali, il mosaicismo di solito esita in sintomi clinici ed anormalità endocrinologiche meno importanti, mentre il fenotipo peggiora progressivamente all’aumentare della severità della polisomia (per esempio nel 49,XXXXY).
Sindrome di Klinefelter caratteristiche 1
Cause della Sindrome di Klinefelter
Queste anomalie cromosomiche trovano una causa materna legata all’ovogenesi nei due terzi dei casi e paterna nel terzo rimanente. Nonostante le cellule germinali primordiali siano presenti alla nascita nei testicoli dei soggetti con sindrome di Klinefelter, queste cellule degenerano molto precocemente; alla pubertà rimangono poche cellule germinali e rari tubuli seminiferi con spermatogenesi completa.
Alla sindrome di Klinefelter è associata una elevata comorbidità in confronto alla popolazione in generale:
- varici,
- trombosi,
- alterazioni della coagulazione,
- diabete di tipo 2,
- fratture ossee,
- problemi neurologici e ritardo mentale
sono le patologie che si presentano con maggiore frequenza.
L’aspettativa di vita è inoltre di 11,5 anni inferiore alla popolazione maschile generale.
Sintomi e diagnosi
Solo il 25% di questi pazienti ricevono una diagnosi in età postnatale e meno del 10% prima della pubertà, quest’ultimo dato assume una rilevanza negativa più significativa in quanto oggi nuove evidenze scientifiche mostrerebbero la possibilità di un più efficace intervento terapeutico, soprattutto sui problemi legati alla spermatogenesi, se la diagnosi fosse più tempestiva.
Considerate le elevate percentuali di pazienti con Sindrome di Klinefelter non diagnosticata o diagnosticata soltanto in età adulta emerge la necessità di riconoscere prima la malattia al fine di garantire un adeguato trattamento e la prevenzione di tutte le eventuali comorbidità e relative complicanze.
L’assetto ormonale dei bambini con Sindrome di Klinefelter non differisce da quello di bambini normali, né per quanto riguarda i livelli sierici di testosterone né per quelli di FSH e LH. In età infantile (0-6 anni), il sospetto può nascere in presenza di criptorchidia, ipospadia, specie se copresenti, di ipogenitalismo, micropene, scroto bifido e di ritardo del linguaggio o ritardo motorio.
Nei bambini identificati con una diagnosi prenatale è importante una valutazione logopedica, anche in assenza di ritardo del linguaggio, per iniziare quanto prima, verso i due -tre anni, un trattamento, ove fosse necessario.
Le varianti della Klinefelter, con polisomia delle X, vengono diagnosticate con maggior facilità a causa delle più importanti alterazioni cliniche e cognitive.
Sindrome di Klinefelter e criptorchidismo
Diversi studi hanno dimostrato che la Sindrome di Klinefelter è la causa genetica più comune di criptorchidismo con una prevalenza tra i criptorchidi dell’1,1%; in questa prospettiva potrebbe essere utile dare l’indicazione di condurre studi citogenetici a tutti i piccoli pazienti che presentano una criptorchidia.
Disturbi cognitivi
I bambini con Klinefelter possono presentare problemi nello sviluppo della socialità e del linguaggio, come anche nella regolazione delle emozioni e del comportamento, comunque i disordini prevalenti sono rappresentati dai disturbi del linguaggio, seguiti dai deficit d'attenzione e dallo spettro di tutti i problemi legati all’autismo.
All’adolescenza la mancanza di sintomi significativi tra ragazzi con Klinefelter e i loro coetanei, rende necessaria, anche per questo gruppo di popolazione, una maggiore attenzione da parte dei familiari, del medico curante e dell’andrologo, quando presente, in modo tale, se sospetto confermato, da indirizzare precocemente questi ragazzi verso strutture sanitarie specializzate e capaci di impostare un corretto iter diagnostico e poi terapeutico.
Sintomi della Sindrome di Klinefelter
I sintomi anche qui sono sempre molto aspecifici e quindi il disordine è sempre sottostimato come del resto nei bambini che negli adulti; ricordiamo ancora una volta che solo un quarto dei maschi con Klinefelter arriva ad avere una diagnosi e poco meno del 10% viene “scoperto” prima della pubertà.
I ragazzi con la Sindrome di Klinefelter iniziano lo sviluppo puberale a un’età simile a quella dei loro coetanei; sono comunque generalmente di statura più alta; il percentile dell’altezza aumenta con l’età e l’incremento staturale precede la pubertà, indicando il coinvolgimento di una componente ormonale non sessuale o di altri fattori nella eziologia dell’alta statura.
È stata dimostrata una correlazione negativa significativa tra i livelli di testosterone sierico e la lunghezza del segmento corporeo inferiore e nel rapporto segmento inferiore/superiore; i soggetti con Klinefelter generalmente sono magri durante il periodo adolescenziale mentre tendono poi a divenire obesi in età adulta. Tale condizione è strettamente associata allo stato di ipogonadismo. È stata comunque dimostrata una resistenza insulinica anche in condizioni di normopeso e quindi un maggior rischio di diabete non insulino-dipendente in questi soggetti.
In questi ragazzi si osserva inoltre una ridotta massa muscolare, soprattutto nella parte superiore del corpo, in particolar modo al torace.
Dopo la pubertà, la presenza di testicoli piccoli e duri e sintomi variabili di deficit androgenico portano all’identificazione di portatori di Klinefelter come spesso succede in presenza di una azoospermia nell’adulto.
Tra i segni fisici caratteristici è da considerarsi soprattutto il volume testicolare ridotto, per diminuite dimensioni della componente tubulare, a far sorgere qualche sospetto; alcuni pazienti (una minoranza) possono sviluppare un ipogonadismo franco con evidenti segni e sintomi di ipovirilizzazione già in età adolescenziale.
A partire dalla fase puberale intermedia i livelli sierici di LH e, soprattutto, di FSH aumentano progressivamente fino a livelli di franca ipergonadotropinemia, con un’aumentata risposta al GnRH test; ancora alla pubertà il testosterone sierico aumenta, raggiungendo valori normali o ai limiti inferiori, assicurando così una normale comparsa dei caratteri sessuali secondari, fatta eccezione per il volume testicolare che non incrementa progressivamente, lasciando così entrambe le gonadi piccole (≤ 4 ml di volume per ogni testicolo) e dure alla palpazione.
Tale disaccoppiamento tra normale progressione dello sviluppo puberale (sviluppo dell’asta, pigmentazione scrotale, sviluppo dei caratteri sessuali secondari) e mancato incremento volumetrico dei testicoli rappresenta il dato clinico più importante ai fini di una diagnosi precoce di Sindrome di Klinefelter in età peripuberale e puberale.
Durante la pubertà, circa il 40% dei pazienti con Sindrome di Klinefelter sviluppa una ginecomastia bilaterale; questo sintomo è il segno di uno squilibrio tra estrogeni e androgeni per eccessiva produzione di estrogeni e/o ridotta secrezione o azione degli androgeni. Nelle prime fasi della pubertà può comparire, o meglio rendersi più evidente, un deficit delle abilità linguistiche e difficoltà di sviluppo del linguaggio che spesso è già presente in età infantile.
Le concentrazioni di testosterone sierico aumentano durante la parte iniziale dell’adolescenza in alcuni pazienti, ma iniziano a decrescere dall’età di 15 anni, risultando al di sotto della norma in circa l’80% dei Klinefelter adulti.
Gli androgeni circolanti sembrano, comunque, essere sufficienti per indurre un normale inizio e progressione della pubertà e lo sviluppo dei caratteri sessuali secondari. L’esatto meccanismo del deficit androgenico non è noto e il grado di disfunzione delle cellule di Leydig è variabile.
Esame obiettivo
L’esame obiettivo a volte permette di evidenziare questo “disaccoppiamento” tra la normale progressione dello sviluppo puberale (sviluppo dell’asta, pigmentazione scrotale, sviluppo dei caratteri sessuali secondari) ed il mancato incremento volumetrico dei testicoli. Tale dato clinico è di estrema rilevanza ai fini di una diagnosi precoce di Sindrome di Klinefelter in età peripuberale e puberale.
Per approfondire:Sperma trasparente e acquoso: tutte le cause
Cosa fare in caso di Sindrome di Klinefelter?
Il ritrovamento di spermatozoi nei testicoli di uomini con Klinefelter ha messo in dubbio l’assunto che essi fossero sempre infertili e pone il quesito se i bambini con tale Sindrome nascono con un numero di spermatogoni severamente ridotto o se c’è un periodo specifico della loro vita nella quale gli spermatogoni vanno in apoptosi massiva.
Dai dati ora disponibili, è ragionevole pensare che la maggior parte degli uomini con Klinefelter alla nascita presentino spermatogoni che vanno poi incontro ad apoptosi massiccia durante la fase puberale quando si osserva l’incremento dei livelli di FSH.
Generalmente l’eiaculato dei soggetti adulti con cariotipo 47, XXY mostra azoospermia, spermatozoi sono stati osservati solo nell’8% dei casi di Klinefelter ma sono anche stati riportati casi eccezionali di paternità spontanea.
La possibilità di valutare la spermatogenesi nella fase iniziale della pubertà fa nascere alcuni problemi di natura etica e pratica, legati alla necessità di ottenere del liquido seminale, attraverso la masturbazione, in ragazzi di età compresa tra i 12 e i 14 anni.
Bisogna considerare sempre la possibile limitata abilità dei ragazzi pre- e puberi nel raccogliere il proprio liquido seminale insieme agli aspetti etici connessi ai problemi di fertilità e alla sessualità dei pazienti di minore età.
In caso di pazienti che non abbiano raggiunto la maggiore età, tuttavia, i dati di letteratura depongono in favore di una buona comprensione e del raggiungimento di una buona consapevolezza di tali tematiche sia da parte del paziente che da parte dei loro genitori, il cui consenso è ovviamente indispensabile. Si deve sempre ottenere un adeguato livello di consenso con i genitori ed il giovane paziente e bisogna essere capaci di porre le giuste istanze legate alla problematica fertilità tenendo sempre presente l’età del ragazzo; se non si ha un eiaculato o non ci sono spermatozoi bisogna, con empatia e abilità discutere, in modo chiaro, di questi aspetti molto delicati.
Tutti queste problematiche vanno affrontate sempre con un atteggiamento aperto e laico, pronti a rispondere a qualsiasi domanda venga poi posta.
Un altro aspetto da approfondire e sul quale mancano studi significativi è la modalità di comunicazione della diagnosi al maschio con la Sindrome di Klinefelter; nel caso di una diagnosi precoce, se il soggetto è minorenne, è sempre indispensabile informare i genitori, dopo di che si deciderà se e come rendere partecipe il giovane paziente. Attualmente non è possibile definire un’età nella quale il soggetto possa dirsi in grado di comprendere il significato di una tale diagnosi, per cui sarà opportuno valutare caso per caso come sia meglio procedere, di concerto con i genitori ed avvalendosi del supporto di altri specialisti (psicologi, medico legali, ed altro ancora).
Per approfondire:Sindrome di Klinefelter e fertilità maschile
In sintesi possiamo dire che nell’adolescente con una Sindrome di Klinefelter oggi è possibile ricorrere alla crioconservazione dello sperma, qualora fossero rilevati spermatozoi nell’eiaculato, ma, come già detto, la raccolta andrebbe eseguita in giovane età, prima della deriva apoptotica delle cellule germinali che si verifica alla pubertà e prima di iniziare il trattamento con androgeni. Sembrano esserci evidenze che gli spermatozoi siano più facilmente trovati, anche tramite una mTESE, in testicoli di ragazzi o maschi più giovani rispetto a pazienti adulti e quindi deve essere presa in considerazione anche la possibile crioconservazione di spermatozoi ottenuti da biopsia testicolare.
Per approfondire:La tecnica Micro-TESE
Una diagnosi precoce e seguire poi questi soggetti fin dalla giovane età permette di instaurare con essi un rapporto che rende il medico esperto un punto di riferimento per dubbi e domande che spesso accompagnano questo genere di diagnosi, limitando in tal modo l’ansia legata alla consapevolezza di essere affetti da una malattia genetica; un ulteriore vantaggio è rappresentato dal fatto che, vista la non del tutto completa conoscenza di questa patologia, studiare subito, in giovane età, i soggetti con Klinefelter potrebbe consentire un avanzamento nelle conoscenze scientifiche circa la storia naturale della malattia e l’efficacia dei trattamenti e degli interventi medici proposti.
La possibilità di una diagnosi precoce presenta numerosi vantaggi che si riflettono sulla possibilità di instaurare precocemente un follow-up, permettendo così di prevenire e/o curare appropriatamente le comorbidità e di fornire al paziente l’adeguato supporto medico e psicologico.
In particolare, l’infertilità può essere risolta con la crioconservazione dello sperma, qualora fossero rilevati precocemente rari spermatozoi nell’eiaculato e la terapia sostitutiva con testosterone può essere intrapresa ai primi segni e/o sintomi di ipogonadismo clinico o laboratoristico.
La somministrazione di testosterone comunque non è generalmente necessaria durante la pubertà poiché la produzione endogena di androgeni è generalmente sufficiente a garantire una normale comparsa e progressione della pubertà.
La terapia sostitutiva con testosterone va iniziata quando richiesta dalla sintomatologia clinica e quando i livelli serici scendono sotto 12 nmol/L di testosterone totale e/o 250 pmol/L di testosterone libero.
In sintesi: cosa fare?
La Sindrome di Klinefelter è una delle più comuni forme di aneuploidie e la più frequente forma d’ipogonadismo e relativa infertilità su base genetica.
Benché la diagnosi sembrerebbe facile (ipogonadismo importante) spesso rimane sottostimata o non trattata.
I bambini e gli adolescenti con Klinefelter posso avere problemi di sviluppo del linguaggio o difficoltà di apprendimento.
La sindrome di Klinefelter può essere associata a diverse comorbidità e ad una aspettativa di vita di circa 11 anni inferiore alla popolazione maschile generale.
La somministrazione di testosterone, quando indicata, è il principale trattamento e, attraverso il recupero chirurgico di spermatozoi “precoce”, alcuni di questi pazienti possono avere la possibilità di diventare genitori.
Altre informazioni
- Klinefelter HF et al. Syndrome characterized by gynecomastia, aspermatogenesis without A-Leydigism, and increased excretion of follicle stimulating hormone. Journal of Clinical Endocrinology 1942;2:615-627.
- Lanfranco F et al. Klinefelter’s syndrome. Lancet 2004;364:273-283.
- Bojesen A et al. Klinefelter syndrome in clinical practice. Nature Clinical Practice Urology 2007;4:192-204.
- Nieschlag E, Behre HM, Wieacker P, Meschede D, Kamischke A, Kliesch S. Störungen im Bereich der Testes. Andrologie. In: Nieschlag E, Behre HM, Nieschlag S, editors. Grundlagen und Klinik der reproduktiven Gesundheit des Mannes. 3rd edition. Heidelberg: Springer; 2009. pp. 199–244.
- Visootsak J et al. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet Journal of Rare Diseases 2006;1:42.
- Tüttelmann F, Gromoll J. Novel genetic aspects of Klinefelter’s syndrome. Mol Hum Reprod. 2010;16:386–395. [PubMed]
- Maiburg M, Repping S, Giltay J. The genetic origin of Klinefelter syndrome and its effect on spermatogenesis. 2012;98:253–260.
- Rochira V. Clinicl Issues in Management of Klinefelter’s Syndrome. In: Meet-the-Professor & Case Management Forum Handouts-ENDO09. The 91st Annual Meeting of the Endocrine Society, Wa-shington, DC, USA, June 10-13. 2009:207-216.
- Wikström AM et al. Klinefelter syndrome in adolescence: onset of puberty is associated with accelerated germ cell depletion. Journal of Clinical Endocrinology and Metabolism 2004;89:2263-2270.
- Swerdlow AJ, Higgins CD, Schoemaker MJ, Wright AF, Jacobs PA. Mortality in patients with Klinefelter syndrome in Britain: a cohort study. J Clin Endocrinol Metab. 2005;90:6516–6522. [PubMed]
- Zitzmann M et al.X-chromosome inactivation patterns and androgen receptor functionality influence phenotype and social characteristics as well as pharmacogenetics of testosterone therapy in Klinefelter patients. Journal of Clinical Endocrinology and Metabolism 2004;89:6208-6217
- Wikström AM et al. Are adolescent boys with Klinefelter syndrome androgen deficient? A longitudinal study of Finnish 47,XXY boys. Pediatric Research 2006;59:854-9.
- Mandok MW, et al. Klinefelter syndrome: the need for early identification and treatment. Clin Pediatr 1991, 30:161-164.
- Ratcliffe S. Long-term outcome in children of sex chromosome abnormalities. Arch Dis Child. 1999;80:192–195. [PMC free article] [PubMed].
- Girardin CM Comparison of adolescents with Klinefelter syndrome according to the circumstances of diagnosis: amniocentesis versus clinical signs. Epub Aug 18 2009; 72(2):98-105.
- Bojesen A et al. Klinefelter syndrome in clinical practice. Nature Clinical Practice Urology 2007;4:192-204.
- Meschede D et al.Klinefelter syndrome. In Oxford Textbook of Endocrinology and Diabetes. Wass JAH & Shalet SM Eds; Male Endocrinology Section Editor Nieschlag E. Oxford University Press 2002;1292-4.
- Lee YS et al.Genital anomalies in Klinefelter’s syndrome. Hormone Research2007;68:150-5.
- Manning MA et al. Diagnosis and management of the adolescent boy with Klinefelter syndrome. Adolescent Medicine 2002;13:367-374.
- Wikström AM et al. Testicular function in Klinefelter syndrome. Horm Res 2008;69:317-26
- Aksglaede L et al. Low semen volume in 47 adolescents and adults with 47,XXY Klinefelter or 46,XX male syndrome. International Journal of Andrology 2009;32:376- 384.37.
- Wattendorf DJ et al.Klinefelter syndrome. American Family Physician 2005;72:2259-2262.
- Bakircioglu ME, Erden HF, Kaplancan T, Ciray N, Bener F, Bahceci M. Aging may adversely affect testicular sperm recovery in patients with Klinefelter syndrome. Urology. 2006;68:1082–1086. [PubMed]
- Van Saen D, Gies I, De Schepper J, Tournaye H, Goossens E. Can pubertal boys with Klinefelter syndrome benefit from spermatogonial stem cell banking? Hum Reprod. 2012;27:323–330. [PubMed]
- Gies I, De Schepper J, Goossens E, van Saen D, Pennings G, Tournaye H. Spermatogonial stem cell preservation in boys with Klinefelter syndrome: to bank or not to bank, that’s the question. Fertil Steril. 98 [PubMed]
- Bastida MG et al.Establishment of testicular endocrine function impairment during childhood and puberty in boys with Klinefelter syndrome. Clinical Endocrinology 2007;67:863-870.
- Zitzmann M, Faber S, Nieschlag E. Association of specific symptoms and metabolic risks with serum testosterone in older men. J Clin Endocrinol Metab. 2006;91:4335–4343. [PubMed]
- Paulis G. Chromosonic Causes of Infertility. In Cavallini G and Beretta G editors.Clinical Management of Male Infertility. Springer International Publishing Heidelberg 2015
Crediti immagini


{kind=link}